微信扫码


下载APP


微信扫码
下载APP
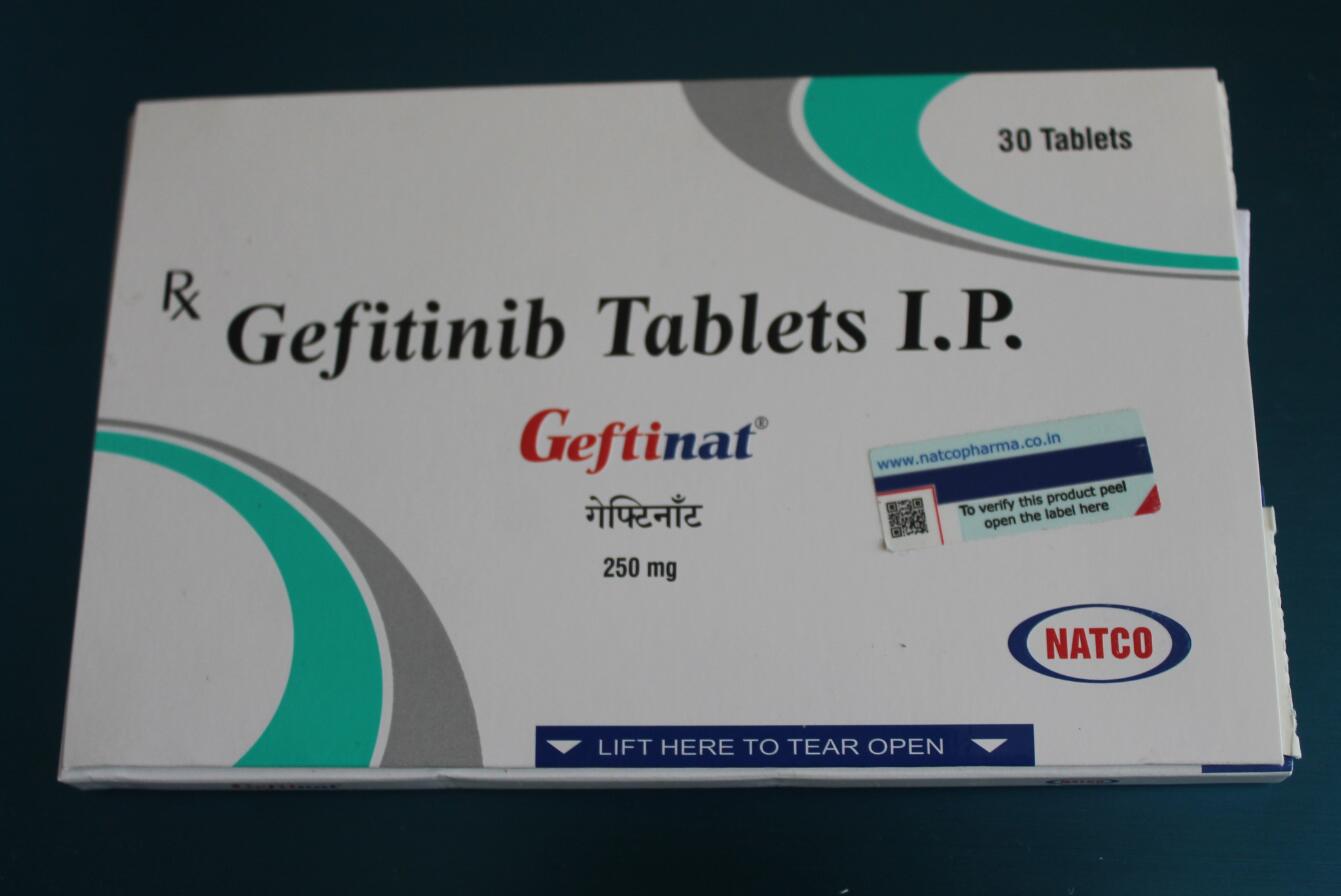
印度Gefitinib Tablets 易瑞沙 印度吉非替尼 伊瑞可
局部晚期或转移性非小细胞肺癌 (NSCLC)
| 数量: | 库存 282 件 |
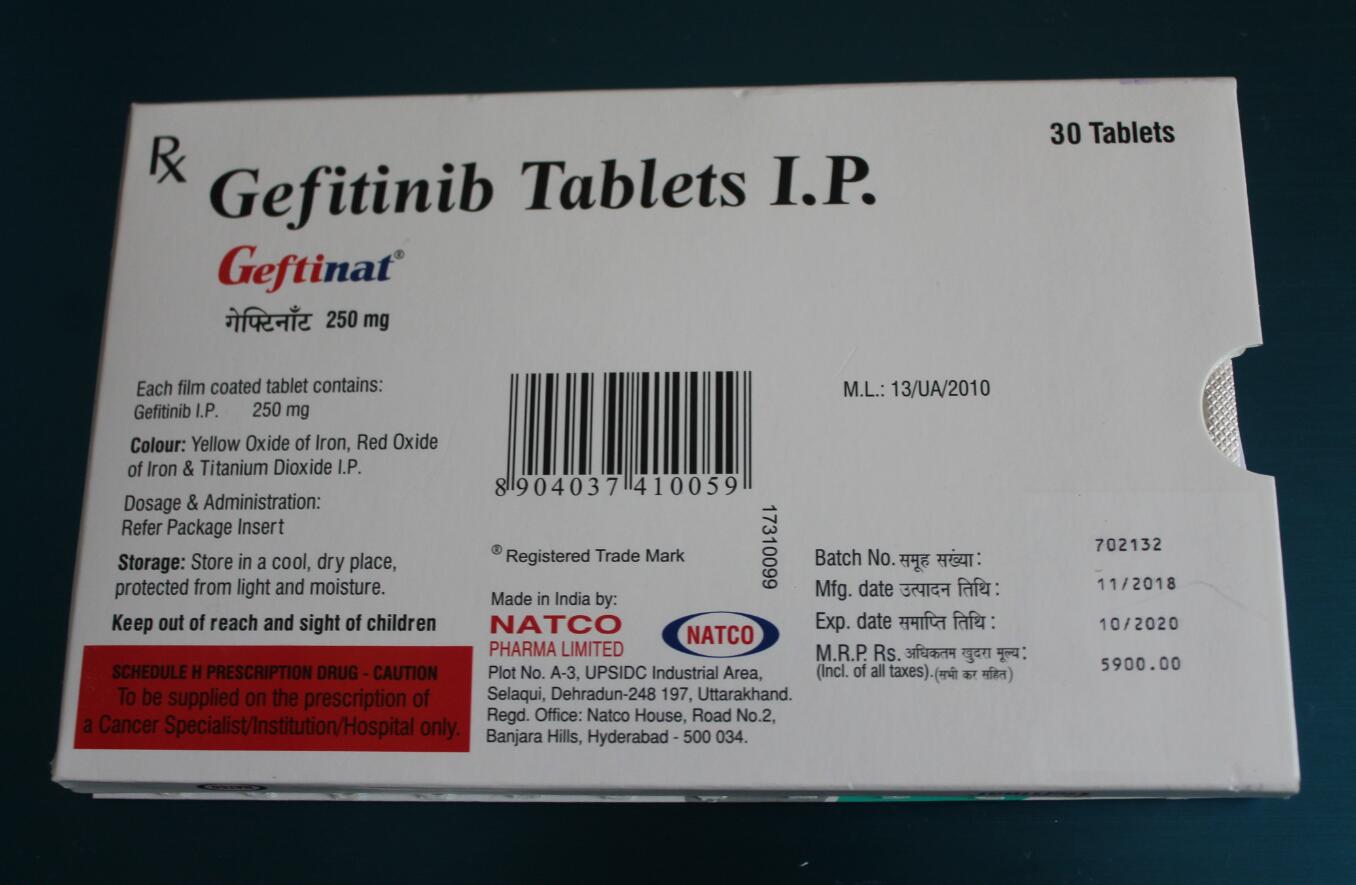
药品名称:印度易瑞沙,印度吉非替尼
有效成分:吉非替尼(Gefitinib)
剂量:250mg
剂型:片剂
包装:30粒/盒
适用症:
本品适用于治疗既往接受过化学治疗的局部晚期或转移性非小细胞肺癌 (NSCLC) 。既往化学治疗主要是指铂剂和多西紫杉醇治疗。
本品对于既往接受过化学治疗的局部晚期或转移性非小细胞肺癌患者的疗效,是基于大规模安慰剂对照临床试验预设亚洲亚组的生存优势 (注:该试验总体人群中未显示改善疾病相关症状和延长生存期) 及中国非对照临床试验的生存数据而确立的。
两个大型的随机对照临床试验结果表明:吉非替尼联合含铂化疗方案壹线治疗局部晚期或转移性NSCLC未显示出临床获益,所以不推荐此类联合方案。
用法用量:本品的推荐剂量为250mg (1片) ,壹日1次,口服,空腹或与食物同服。
如果有吞咽困难,可将片剂分散于半杯饮用水中 (非碳酸饮料) 。不得使用其他液体。将片剂丢入水中,无需压碎,搅拌至完全分散(约需10分钟),即刻饮下药液。以半杯水冲洗杯子,饮下。也可通过鼻-胃管给予该药液。
无需因下述情况不同调整给药剂量:年龄、体重、性别、种族、肾功能、因肝转移而引起的中至重度肝功能损害。
剂量调整:当患者出现不能耐受的腹泻或皮肤不良反应时,可通过短期暂停治疗(最多14天)解决,随后恢复每天250mg的剂量。
目前尚无本品用于儿童或青少年患者安全性与疗效的数据,故不推荐使用。
百度百科您身边的百科全书
本品的推荐剂量为250mg(1片),一日1次,口服,空腹或与食物同服直到出现疾病进展或不能耐受的毒性。如果漏服本品一次,应在患者记起后尽快服用。如果距离下次服药时间不足12小时,则患者不应再服用漏服的药物。患者不可为了弥补漏服的剂量而服用加倍的剂量(一次服用两倍剂量)。
当不能整个片剂给药时,例如患者只能吞咽液体,可将片剂分散于水中。片剂应分散于半杯饮用水中(非碳酸饮料)无需压碎,搅拌至完全分散(约需15分钟),即刻饮下药液。以半杯水冲洗杯子,饮下洗液。也可通过鼻胃管给予该药液。
药物不良反应所致剂量调整
出现以下任何一种情况的患者应暂停吉非替尼给药(长达14天):
以下情况需永久终止吉非替尼治疗(见【注意事项】):
药物相互作用剂量调整
CYP3A4强诱导剂
如果未出现重度药物不良反应,吉非替尼日剂量可增加至500mg,中止强效CYP3A4诱导剂给药后7天,重新开始吉非替尼250mg给药(见【药物相互作用】、【药代动力学】)。
CYP3A4抑制剂
强效CYP3A4抑制剂(如酮康唑和伊曲康唑)能降低吉非替尼代谢,增加其血浆浓度。吉非替尼与强效CYP3A4抑制剂合并用药时,应监测不良反应(见【药物相互作用】)。
肝功能损伤
肝硬化所致中度至重度肝功能损伤(ChildPughB或C)的患者吉非替尼血浆浓度增高。应密切监控这些患者的不良事件。肝转移导致天冬氨酸转氨酶(AST)、碱性磷酸酶或胆红素升高的患者其血药浓度未见升高(见【药代动力学】、【注意事项】)。
肾功能损伤
肌酐清除率>20ml/分的肾功能损伤患者无需调整剂量。因肌酐清除率≤20ml/分的患者的数据有限,因此,这些患者用药时应谨慎(见【药代动力学】)。
安全性特征概述
来自ISEL、INTEREST和IPASS三项Ⅲ期临床试验(包括2462例接受吉非替尼250mg每日一次单药治疗的患者)的汇总数据集中,最常见(发生率20%以上)的药物不良反应(ADRs)为腹泻和皮肤反应(包括皮疹、痤疮、皮肤干燥和瘙痒),一般见于服药后的第一个月内,通常是可逆性的。
大约10%的患者出现严重的药物不良反应(按照美国国立癌症研究所[NCI]通用毒性评价标准[CTC]3或4级)。因ADR停止治疗的患者有约3%。
接受吉非替尼治疗的2462例患者中,1.3%患者发生ILD或类似ILD的药物不良反应(如,肺浸润、肺炎、急性呼吸窘迫综合症或肺纤维化),其中,0.7%为3级或更高级,3例为致死性不良反应。
这些研究中剔除了具有间质性肺疾病、药物诱导的间质性疾病、需甾体类药物治疗的放射性肺炎或具有临床意义的活动性间质性肺疾病病史的患者。
不良事件列表
表1给出了临床试验中和上市后报告的安全性概况,列出了相关不良反应发生频率的分类,这些发生率是基于III期临床试验ISEL、INTEREST和IPASS的汇总数据集中报告的相应不良事件的发生率。
频率的分类没有考虑对照组报告的不良事件发生率,也未考虑研究者判断的与试验药物相关性。与实验室检查异常相关的不良反应的发生率,是基于相关的化验指标与基线相比变化程度达到或超过2个CTC级别的患者。
各身体系统发生的不良反应按发生频率以降序排列。不良反应发生频率定义为:十分常见:>10%;常见:>1%且<10%;偶见:>0.1%且<1%;罕见:>0.01%且<0.1%;十分罕见:<0.01%。
表1:按照系统/器官和发生率列出不良反应

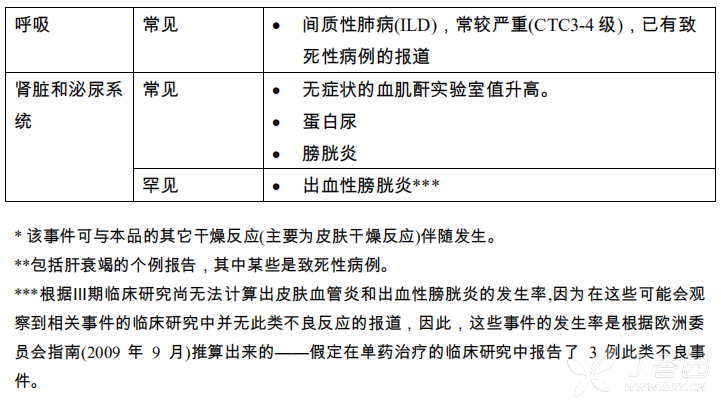
间质性肺病
一项双盲的III期临床研究中(ISEL),比较本品加最佳支持治疗(BSC)与安慰剂加BSC用于在既往接受过一个或两个化疗方案,且对最近的治疗无效或不耐受的局部晚期NSCLC患者(1692例患者),间质性肺病型事件的发生率在总体人群中相似,两治疗组均为约1%。
绝大多数报道的间质性肺病型事件均来自亚裔人群,接受本品或安慰剂治疗的亚裔人群患者中,间质性肺病的发生率相似,分别为约3%和4%。一例间质性肺病型事件导致死亡,为接受安慰剂治疗的患者。
在日本的上市后药物监测研究中(3350名患者),报道的接受本品治疗的患者间质性肺病型事件的发生率为5.8%。
一项在日本的非小细胞肺癌患者中进行的流行病学病例对照研究中,12周随访时间内间质性肺病的累积发生率的原始数据(未根据患者特征的不均衡而调整),接受本品的患者为4.0%,接受化疗的患者为2.1%;与接受化疗相比,接受本品治疗发生间质性肺病的校正比值比为3.2(95%的可信区间为1.9-5.4)。
相对于化疗而言,使用本品发生间质性肺病的风险升高,这主要见于治疗的前四周(校正比值比为3.8,95%的可信区间为1.9-7.7),此后的相对风险较低(校正比值比为2.5,95%的可信区间为1.1-5.8)。
在一项比较本品与卡铂/紫杉醇二联化疗用于一线治疗选择性的晚期NSCLC亚洲患者(1217例)的III期开放临床试验中(IPASS),ILD型事件的发生率在吉非替尼治疗组为2.6%,卡铂/紫杉醇治疗组为1.4%。
在该项临床研究的中国患者中,吉非替尼治疗组发生1例ILD型事件,为0.5%,卡铂/紫杉醇治疗组发生4例,为2.2%。
当考虑本品用于晚期或转移性NSCLC患者的治疗时,应对所有患者的肿瘤组织进行EGFR突变检测。确定具有EGFR基因敏感突变的患者推荐本品治疗。如果肿瘤标本不可评估,则可使用从血液(血浆)标本中获得的循环肿瘤DNA(ctDNA)。
只能使用经论证可用于测定肿瘤或ctDNA的EGFR突变状态的检测方法,检测方法须稳定、可靠并且灵敏,以避免出现假阴性或假阳性的测定结果。
间质性肺病
观察到接受本品治疗的患者发生间质性肺病,可急性发作,有死亡病例报告(见【不良反应】)。如果患者呼吸道症状恶化,如呼吸困难,咳嗽,发热,应中断本品治疗,立即进行检查。当证实有间质性肺病时,应停止使用本品,并对患者进行相应的治疗。
一项在日本进行的流行病学病例对照研究中,对接受本品或化疗的3159名非小细胞肺癌患者进行为期12周的随访,确定了以下出现间质性肺病的高风险因素(不考虑患者接受的本品还是化疗):吸烟,较差的体力状态(PS≥2),在CT扫描上正常肺组织覆盖范围≤50%,距非小细胞肺癌诊断时间较短(<6个月),原有间质性肺炎,年龄较大(≥55岁),伴有心脏疾病。两治疗组中发展为ILD的患者,如具有以下危险因素则死亡率增加:吸烟、在CT扫描上正常肺组织减少≤50%、原有间质性肺炎、年龄较大(≥65岁),病灶与胸膜粘连(≥50%)。
肝毒性
已观察到肝功能检查异常(包括丙氨酸氨基转移酶升高,天门冬氨酸氨基转移酶升高,胆红素升高)(见【不良反应】),偶见有表现为肝炎。已有肝衰竭的个例报告,其中某些是致死性病例。因此,建议定期检查肝功能。肝转氨酶轻中度升高的患者应慎用本品。如果肝转氨酶升高加重,应考虑停药。
严重持续的腹泻
当患者出现重度或持续性腹泻、恶心、呕吐或厌食症状时,应告诫其即刻就医,因为这些症状均可能间接引起脱水。这些症状应按临床指证进行处理。
消化道穿孔
服用本品治疗的患者中已有消化道穿孔的报道,涉及的大多数患者本身包含其他已知的风险因素(如,同时服用类固醇药物、非甾体类抗炎药;消化道基础疾病、溃疡、年龄、吸烟史、穿孔部位的肠道转移肿瘤)。
眼部症状
出现任何提示角膜炎的症状或体征(如急性或加重的:眼部炎症、流泪、光敏感、视力模糊、眼部疼痛和/或眼部发红)的患者应立即转诊至眼科专科医生处。
如确诊为溃疡性角膜炎,则应中断本品治疗;如果症状无缓解,或症状在再次服用吉非替尼时复发,则应考虑永久性终止本品治疗。
大疱性和脱皮性皮肤病
服用本品治疗的患者有报告发生大疱性病症,包括中毒性表皮坏死松懈症、Stevens-Johnson综合症和多形性红斑。接受吉非替尼治疗的2462例患者中,2(0.08%)例患者报告多形性红斑和大疱性皮炎。如果患者出现重度大疱性、发疱或脱皮病症,应中断或中止吉非替尼治疗。
其它
在本品的临床试验中有脑血管事件的报告,但与本品的关系未确定。
在一项对儿科患者进行本品和放疗治疗的I/II期临床研究中,45名入选患者(这些患者为新诊断出脑干神经胶质瘤或未完全切除的幕上恶性神经胶质瘤)中,发生4例(1例死亡)中枢神经系统出血。
在一项单用本品治疗的临床研究中,一位患有室管膜瘤的儿童也出现了中枢神经系统出血。没有证据证明接受本品治疗的成年NSCLC患者的脑出血风险增高。
对驾驶及操纵机器能力的影响
在本品治疗期间,可出现虚弱的症状,出现这些症状的患者在驾驶或操纵机器时应给予提醒。
对人肝微粒体进行的体外试验证实,吉非替尼主要通过肝细胞色素P-450系统的CYP3A4代谢。所以吉非替尼可能会与诱导,抑制或为同一肝药酶代谢的药物发生相互作用。动物研究表明吉非替尼很少有酶诱导作用,体外研究显示吉非替尼可有限地抑制CYP2D6。
以下列出了与吉非替尼产生或可能产生有临床意义的药物相互作用的药物或药物类别:
影响吉非替尼的药物
已证明的相互作用
抑制CYP3A4的药物
在健康志愿者中将吉非替尼与伊曲康唑(一种CYP3A4抑制剂)合用,吉非替尼的平均AUC升高80%。由于药物不良反应与剂量及暴露量相关,该升高可能有临床意义。虽然未进行与其他CYP3A4抑制剂相互作用的研究,但这一类药物如酮康唑,克霉唑,Ritonovir同样可能抑制吉非替尼的代谢。
升高胃pH值的药物
在一项健康志愿者中进行临床研究,表明与剂量达到能明显持续升高胃pH至≥5的雷尼替丁合用,可使吉非替尼的平均AUC降低47%,这可能降低吉非替尼疗效。
利福平
在健康志愿者中将吉非替尼与利福平(已知的强CYP3A4诱导剂)同时给药,吉非替尼的平均AUC比单服时降低83%。
理论上可能有相互作用的药物
其他CYP3A4诱导剂
诱导CYP3A4活性的物质可增加吉非替尼的代谢并降低其血浆浓度。因此,与CYP3A4诱导剂(如苯妥因、卡马西平、巴比妥类或圣约翰草)合用可降低疗效。
吉非替尼对其他药物的作用
已证明的相互作用
通过CYP2D6代谢的药物
在一项临床试验中,吉非替尼与美托洛尔(一种CYP2D6酶底物)合用,使美托洛尔的暴露量升高35%,这被认为不具有临床相关性。吉非替尼与其他由CYP2D6代谢的药物同服,可能会升高后者的血药浓度。
理论上可能有相互作用的药物
华法林
虽然迄今尚未进行正规的药物相互作用研究,在一些服用华法林的患者中报告了INR增高和/或出血事件。服用华法林的患者应定期监测其凝血酶原时间或INR的改变(见【注意事项】)。
长春瑞滨
在II期临床研究中,将本品与长春瑞滨同时服用,显示本品可能会加剧长春瑞滨引起的中性白细胞减少作用。
IFUM研究
一项多中心、单组、开放性临床研究证实吉非替尼(IRESSA)作为一线治疗药物对报告EGFR外显子19缺失或L858R置换突变的转移性NSCLC患者的有效性和安全性。
共106例未接受过治疗的转移性EGFR突变阳性NSCLC患者接受250mgIRESSA给药,每日一次,直至出现疾病进展或不耐受毒性。主要疗效结果指标为盲态独立中心审核委员会(BICR)和研究者按RECISTv1.1标准评价的客观缓解率(ORR)。试验还包括其他结果测量指标缓解持续时间(DOR)。
根据临床试验分析预期测定结果,合格患者需具有EGFR外显子19缺失或L858R、L861Q、或G719X置换突变,且肿瘤标本无T790M或S768I突变或外显子20插入。采用therascreen®EGFRRGQPCR试剂盒对87例患者的肿瘤样本进行回顾性检测。
研究人群特征包括:中位年龄65岁,年龄为75岁或以上(25%),年龄小于65岁(49%),白人(100%),女性(71%),从不吸烟者(64%),WHOPS0(45%),WHOPS1(48%),WHOPS2(7%)和组织学腺癌(97%)。60例患者报告外显子19缺失(65%),29例患者报告L858R置换突变(31%),2例患者各报告肿瘤标本出现L861Q或G719X置换突变。
中位疗程为8.0个月,有效性结果总结如下表2。
表2疗效总结
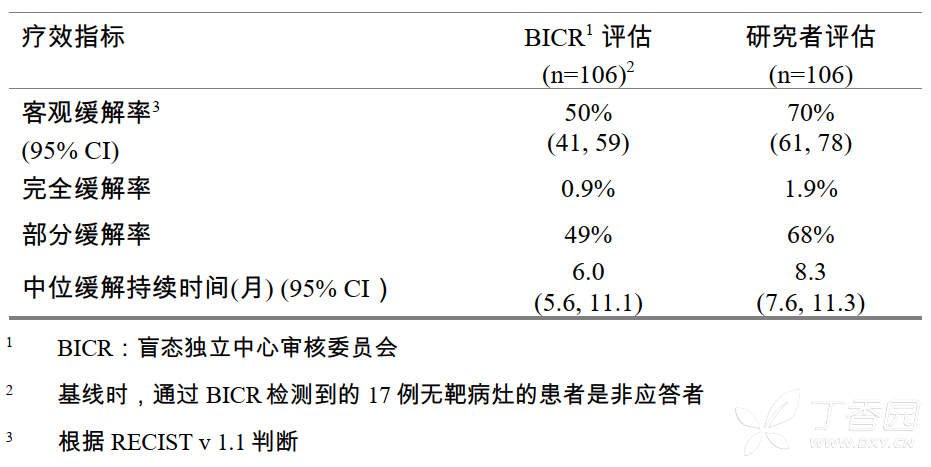
报告EGFR外显子19缺失和外显子21L858R置换突变的患者的缓解率相似。在肿瘤标本出现G719X置换突变的患者中观察到2例部分缓解,最小缓解持续时间分别为2.8个月和5.6个月。在2例肿瘤标本出现L861Q置换突变的患者中,1例患者还达到部分缓解,最小缓解持续时间为2.8个月。
循环肿瘤DNA(ctDNA)
在IFUM试验中,采用TherascreenEGFRRGQPCR试剂盒(Qiagen),对来源于血浆的ctDNA和肿瘤组织样本进行了突变状态评估。1060例经筛查的患者中,有652例患者的ctDNA和肿瘤组织样本均可评估。
肿瘤组织和ctDNA均突变阳性患者的客观缓解率(ORR)为77%(95%CI:66%〜86%),而仅为肿瘤组织突变阳性的患者ORR为60%(95%CI:44%,74%)。
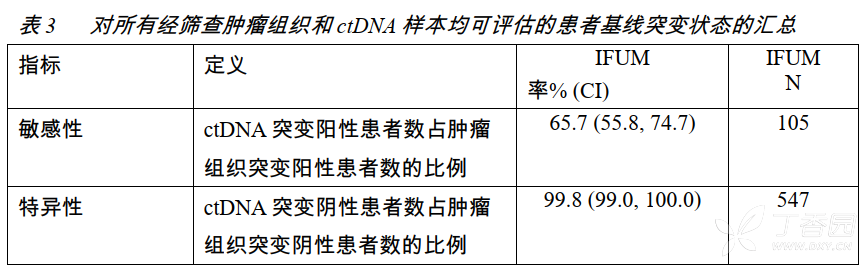
这些数据与IPASS(Goto2012)试验预先计划的探索性日本亚组分析的结果一致。在IPASS研究中采用EGFR突变检测试剂盒(DxS)(N=86),使用来源于血清而非血浆的ctDNA,进行EGFR基因突变分析。该项研究中,敏感性为43.1%,特异性为100%。
INFORM研究:
一项在中国27个中心进行的,在局部晚期或转移性非小细胞肺癌患者中应用吉非替尼维持治疗的随机、安慰剂对照、平行组临床研究。该研究探讨了吉非替尼对比安慰剂在局部晚期或转移性非小细胞肺癌患者一线标准含铂方案化疗后维持治疗中的疗效、安全性和耐受性。患者(≥18岁,IIIB-IV期NSCLC,PS评分0-2)完成4周期一线含铂双药化疗后无进展或不可耐受毒副作用,按1:1比例随机分入吉非替尼组(250mg/日)或安慰剂组。主要研究终点是无进展生存期(PFS)。次要终点包括总生存(OS),客观缓解率(ORR),疾病控制率(DCR),症状改善率及耐受性。
共296名患者(n=148吉非替尼组,n=148安慰剂组)参与随机分组(入组时间为2008年9月26日-2009年8月10日),两组人群的人口学和疾病特征相匹配(见表4)。PFS数据截止至2011年1月24日,患者中91%疾病进展,58%死亡。
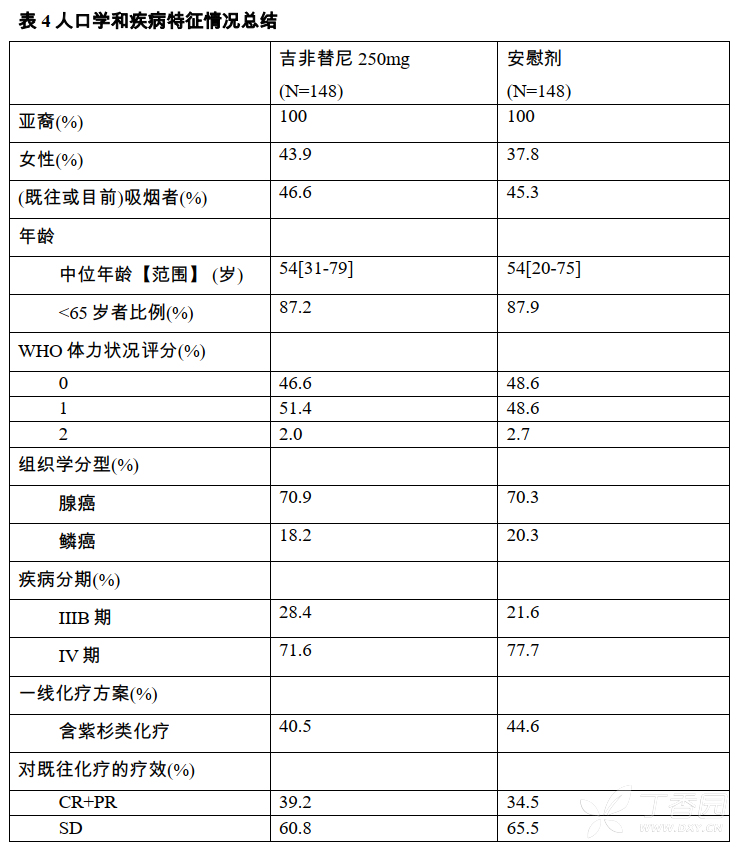
结果显示,在中国局部晚期或转移性非小细胞肺癌患者中吉非替尼维持治疗组较安慰剂组的无进展生存期(PFS)显著延长,具体疗效结果见表5。
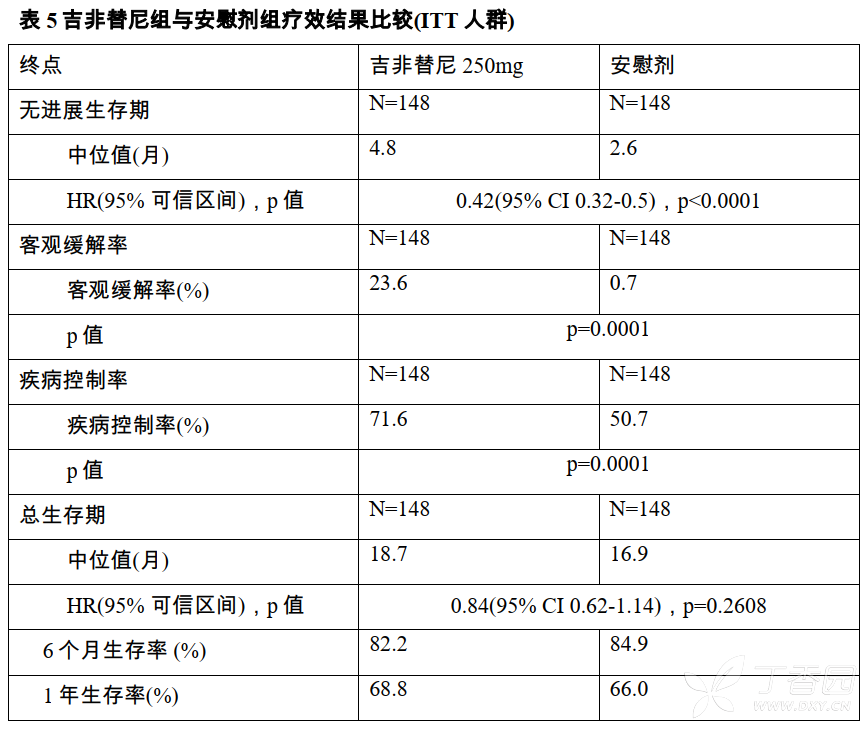
共79例患者(27%)进行了肿瘤组织EGFR突变检测,其基线特征与ITT人群相似,EGFR突变阳性患者共30例(38.0%),两组EGFR敏感突变型/野生型(例)分别为15/25vs15/24。两组EGFR突变阳性患者中位PFS(月)16.6vs2.8,HR=0.17(95%CI0.07-0.42);EGFR野生型患者中位PFS(月)2.7vs1.5,HR=0.86(95%CI0.48-1.51)。
IPASS研究:
一项在亚洲,包括中国大陆,中国香港,印度尼西亚,日本,马来西亚,菲律宾,新加坡,中国台湾,泰国9个国家和地区,87个中心进行的III期临床试验中入组了1217例晚期(IIIB或IV期)NSCLC患者,这些患者需符合如下条件:肿瘤组织学类型为腺癌,既往轻度吸烟(停止吸烟>15年,吸烟<10包/年)或从未吸烟,且既往没有接受过化疗。
试验证明吉非替尼在无进展生存期(PFS)的指标上优于卡铂(AUC5.0或6.0)/紫杉醇(200mg/m2)。这一结果随时间而改变,开始的时候显示卡铂/紫杉醇组更有益,随后显示吉非替尼组更有益,造成PFS结果不同的潜在原因是EGFR突变状态。EGFR突变状态是吉非替尼和卡铂/紫杉醇相比的疗效的强有力的预测性生物标志物。
吉非替尼组的客观缓解率(ORR,采用RECIST评估(完全缓解[CR]+部分缓解[PR]),以下同)比卡铂/紫杉醇组更优。与卡铂/紫杉醇组相比,明显有更多的接受吉非替尼治疗的患者获得了有临床意义的生活质量的提高。
对生物标志物数据进行了预先计划的探索性分析。共有437例患者具有可评价的EGFR突变分析数据。
在EGFR突变阳性的患者中,吉非替尼组的PFS显著长于卡铂/紫杉醇组,而在EGFR突变阴性的患者中,卡铂/紫杉醇组的PFS显著长于吉非替尼组。
使用DxSEGFR29突变检测试剂盒检测,如果使用突变阻断扩增系统(ARMS)方法,检测到29个EGFR突变中至少一个突变时,患者即被视为EGFR突变阳性(M+)。如果标本分析成功,但在29个EGFR突变中,没有检出任何一个EGFR突变,则认为患者突变阴性(M-)。EGFR突变状态不明的亚组人群,其PFS结果(吉非替尼组的风险比:
0.68;95%置信区间:0.58~0.81;P<0.0001)与总人群相似。
在EGFR突变阳性的患者中,吉非替尼组的ORR优于卡铂/紫杉醇组。在EGFR突变阴性的患者中卡铂/紫杉醇组的ORR优于吉非替尼组。IPASS临床研究中,吉非替尼组与卡铂/紫杉醇组相比较的疗效结果见表6。

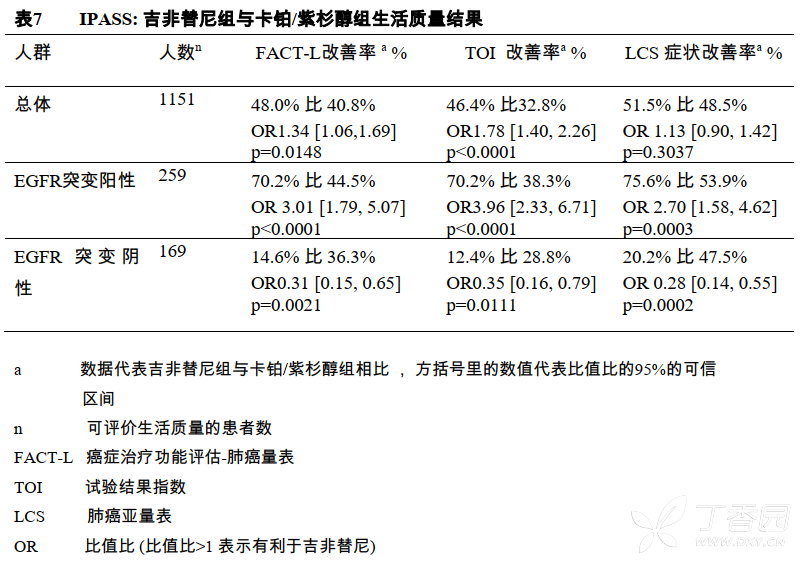
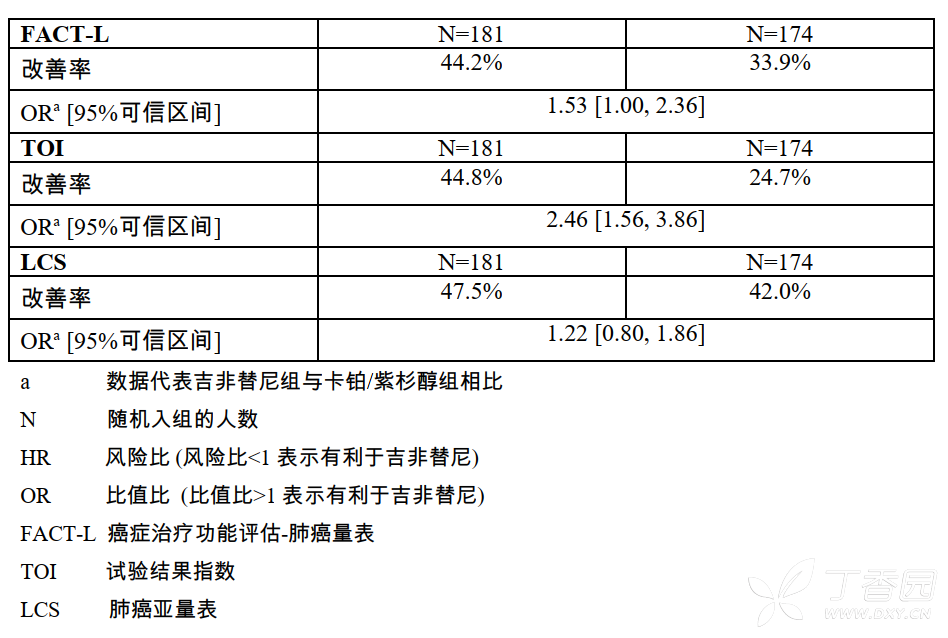
在EGFR突变阳性的患者中,与卡铂/紫杉醇组相比,明显有更多吉非替尼治疗的患者获得生活质量和肺部症状的改善。
在EGFR突变阴性的患者中,与吉非替尼组相比,明显有更多接受卡铂/紫杉醇组的患者获得生活质量和肺癌症状的改善。IPASS临床研究中,吉非替尼组与卡铂/紫杉醇组生活质量相比较的数据见表7。
IPASS研究中国亚组结果
在IPASS临床研究中,来自中国大陆的20个研究中心,372名患者(为研究总人数的31%)随机分为吉非替尼治疗组及卡铂/紫杉醇治疗组。研究的主要终点PFS在中国患者中的结果与总体的结果保持一致(不同国家和地区之间的交互检验P=0.4265)。具体临床研究结果见表8。
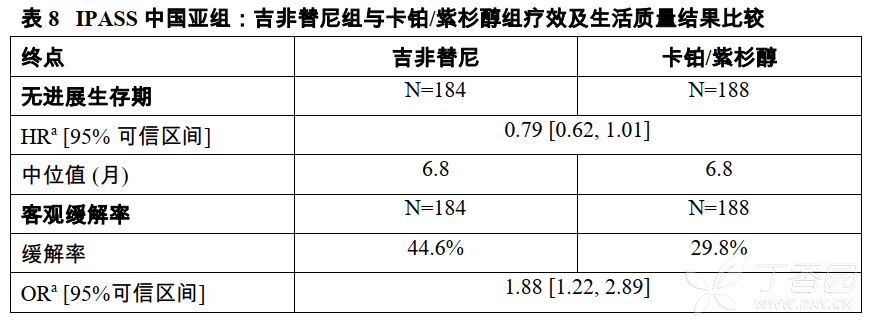
在该研究中,中国患者提供肿瘤标本供检测的比例与总体人群相比较低。共有87例中国患者具有可评价的EGFR突变分析数据[EGFR突变阳性为39例,阴性为48例]。
在EGFR突变阳性的患者中,无进展生存的风险比有利于吉非替尼(HR=0.648,95%CI: 0.264, 1.591),在EGFR突变阴性的患者中,无进展生存的风险比有利于卡铂/紫杉醇(HR=3.552, 95%CI: 1.724, 7.318)。
对患者总生存期的分析结果表明,在总体研究人群中,吉非替尼治疗组的总生存期与卡铂/紫杉醇组相比无显著的统计学差异(风险比:0.901,95%置信区间:0.793~1.023;P=0.1087)。中位总生存期:吉非替尼治疗组为18.8个月,卡铂/紫杉醇治疗组为17.4个月。
根据EGFR突变状态,对亚组患者的总生存期进行分析,结果表明,吉非替尼治疗组与卡铂/紫杉醇治疗组相比,无EGFR突变阳性(风险比:1.002;95%置信区间:0.756 ~1.328;中位总生存期分别为21.6个月和21.9个月)还是EGFR突变阴性(风险比:1.181;95%置信区间:0.857~1.628;中位总生存期分别为11.2个月和12.7个月)的患者,其总生存期并无显著差异;突变状态不明的亚组患者其总生存期结果与总体人群一致(风险比:0.818;95%置信区间:0.696~0.962;中位总生存期分别为:18.9个月和17.2 个月)。
在IPASS试验中,对于表皮生长因子受体(EGFR)基因具有敏感突变的且之前未接受任何治疗的局部晚期或转移性非小细胞肺癌患者而言,使用吉非替尼治疗与卡铂/紫杉醇治疗相比,前者能延长患者的无进展生存期、获得更高的总缓解率、更好的生活质量和更高的症状缓解率;但两者在总生存期上并无显著差异。
INTEREST研究:
在一项纳入24个国家和地区,149个中心进行的III期临床试验,共入组了1466例既往接受过以铂类为基础的化疗,并适合进行进一步化疗的局部晚期或转移性NSCLC患者。(803例患者[55%]来自欧洲, 308例[21%]来自亚洲, 223例[15%]来自北美洲,以及132例[9%]来自美洲的南部和中部)。
患者的中位年龄为61岁(范围从20岁到84岁),34.9%的患者为女性,74.4%为高加索人种,22.0%为亚裔人种。曾经接受过一个,两个和三个化疗方案的患者分别为1229例,235例和2例;所有患者都接受过铂类化疗,其中271例患者曾接受过紫杉醇治疗。
该试验证实吉非替尼治疗的总体生存期不劣于多西他赛(75mg/2)。吉非替尼组的无进展生存期(PFS)和客观缓解率(ORR)也与多西他赛组相似。
与多西他赛组相比,更多接受吉非替尼治疗的患者获得了有临床意义的生活质量的改善。两治疗组出现肺癌症状的改善的比例相似。INTEREST临床研究结果见表9。
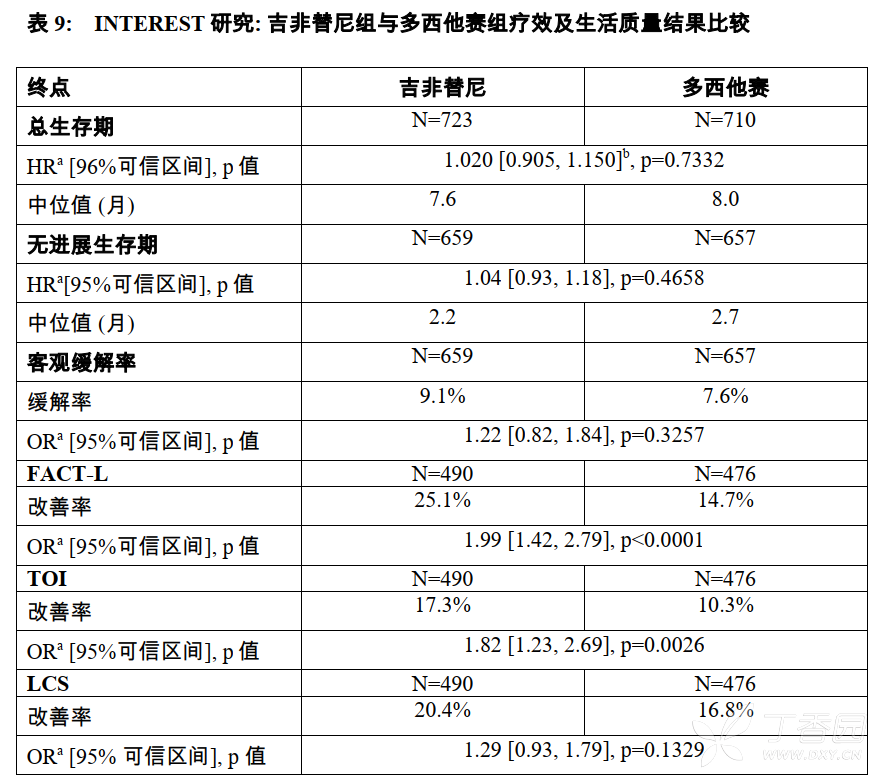
a 数据代表吉非替尼组与多西他赛组相比
b 可信区间完全在非劣效性界值1.154之下
N 随机入组的人数
HR 风险比(风险比<1表示有利于吉非替尼)
OR 比值比(比值比>1表示有利于吉非替尼)
FACT-L癌症治疗功能评估-肺癌量表
TOI 试验结果指数
LCS 肺癌亚量表
预设的主要协变量分析纳入了174例EGFR基因高拷贝数的患者的生存期,其结果显示吉非替尼组并未优于多西他赛。两治疗组高EGFR基因拷贝数的患者的生存结果相似(HR 1.087, 95% CI 0.782~1.510, p=0.6199,中位值8.4比7.5个月)。
对生物标志物数据进行了预先计划的探索性分析。共有297例患者具有可评价的EGFR突变分析数据[EGFR突变阳性为44例,阴性为253例]。在EGFR突变阳性的患者中,总生存的风险比为0.832(95%CI:0.414-1.670),在EGFR突变阴性的患者中,总生存的风险比为1.015(95%CI:0.776-1.327)。
INTEREST研究中国亚组结果
在INTEREST临床研究中,入组了222例中国患者,(为总研究人数的15%)。中国患者的总生存结果与研究的总体结果保持一致(不同国家之间的交互检验P=0.7831)。INTEREST研究中,中国亚组的疗效及生活质量结果见表10。
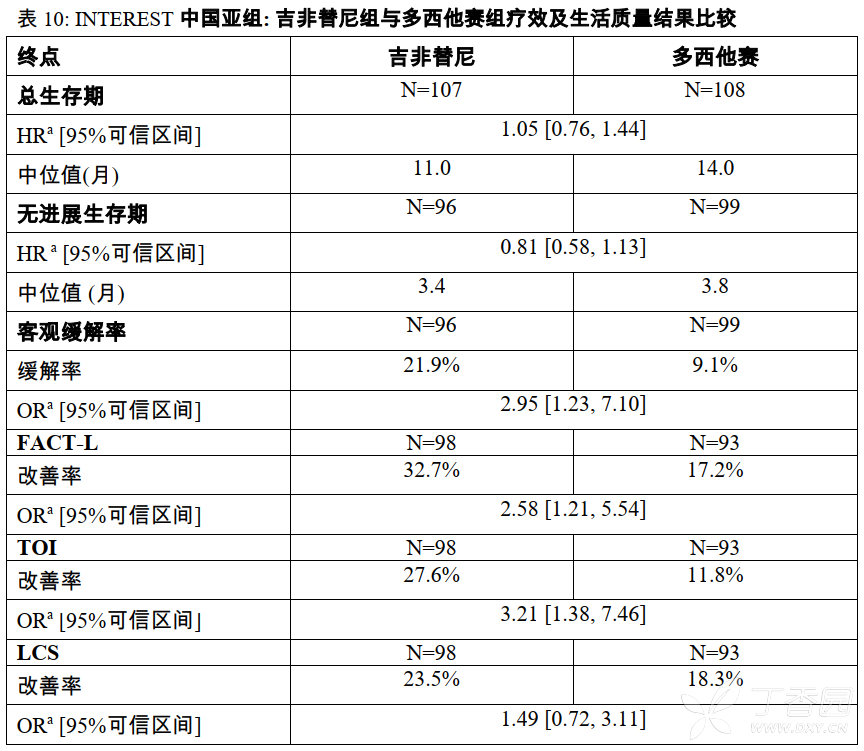
a 数据代表吉非替尼组与多西他赛组相比
N 随机入组的人数
HR 风险比(风险比<1表示有利于吉非替尼)
OR 比值比(比值比>1表示有利于吉非替尼)
FACT-L 癌症治疗功能评估-肺癌量表
TOI 试验结果指数
LCS 肺癌亚量表
在该研究中共有37例中国患者的EGFR突变状态已知:EGFR突变阳性为14例,阴性为23例。由于EGFR突变状态已知的中国患者人数较少,因此没有对疗效变量进行探索性分析。
ISEL研究:
在一项包括欧洲,亚洲,中部和南部美洲,澳大利亚及加拿大等28个国家,210个中心进行的III期双盲研究中,入组1692例既往接受过1次或2次化疗方案,且最近一次化疗耐药或不耐受的晚期非小细胞肺癌患者,比较接受吉非替尼加最佳支持治疗(BSC)或接受安慰剂加最佳支持治疗。
患者的中位年龄为61岁(范围从28岁到90岁),其中32.7%的患者为女性,75.3%的患者为高加索人种,20.2%的患者为亚裔人种。未接受过化疗,曾接受过一个,两个和三个及以上化疗方案的患者分别为2例,823例,847例和20例;接受过铂类化疗的患者有1623例,其中462例患者曾接受过多西他赛治疗。
结果,吉非替尼未能达到有统计学意义的延长总体人群的生存期(HR0.89,CI0.77~1.02,p=0.09,吉非替尼组中位生存期为5.6月,安慰剂组为5.1个月),或有统计学意义的延长腺癌患者的生存期(HR0.84,CI0.68~1.03,p=0.09,吉非替尼组和安慰剂组的中位值分别为6.3比5.4个月)。
预先设定的亚组分析显示,在亚裔人群中,接受吉非替尼治疗组比安慰剂组的生存期延长,有统计学意义(HR=0.66,CI0.48~0.91,p=0.01,中位值9.5比5.5个月),非吸烟的患者接受吉非替尼治疗的生存期也比安慰剂组显著延长(HR=0.67,CI0.49~0.92,p=0.01,中位值8.9比6.1个月)。
探索性分析EGFR基因拷贝数的数据显示,与安慰剂相比,吉非替尼对生存期的治疗作用在EGFR基因高拷贝的患者高于EGFR基因低拷贝的患者(交互作用的p值=0.0448)。在EGFR基因高拷贝的患者中,吉非替尼比安慰剂的风险比(HR)为0.61(N=114;95%CI0.36~1.04,p=0.067),在EGFR基因低拷贝的患者,其风险比为1.16(N=256;95%CI0.81~1.64,p=0.42)。对没有检测EGFR基因拷贝数的患者,正如预期的情况一样,HR与总体人群相似(N=1322,HR=0.85,CI0.73~0.99,p=0.032)。
在中国进行的注册临床研究:
在中国的5个研究中心进行了临床研究,以评估吉非替尼片250mg/日在既往接受过化学治疗的晚期非小细胞肺癌患者中的客观缓解率。
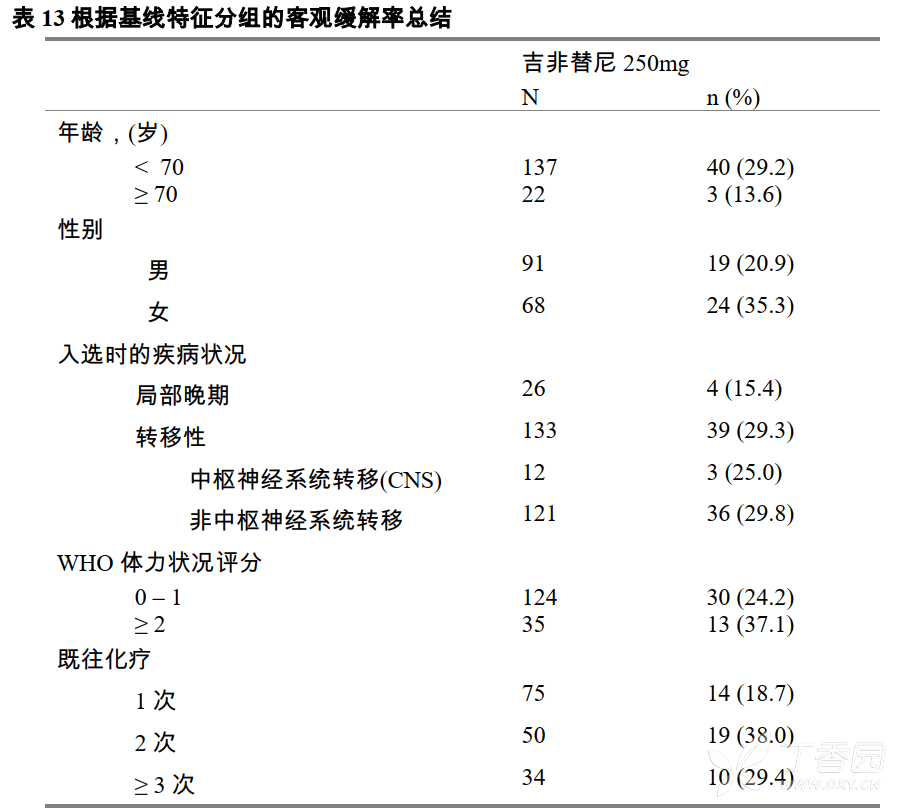
共有159名受试者至少服用了一次吉非替尼片250mg,受试者的人口学和疾病特征情况见表11。其中在入选前曾接受过1个化疗方案治疗的受试者有75名(47.2%),2个及3个以上(含3个)化疗方案治疗的受试者分别为50名(31.4%)和34名(21.4%)。对于159名受试者(意向性治疗人群集)进行了有效性分析。

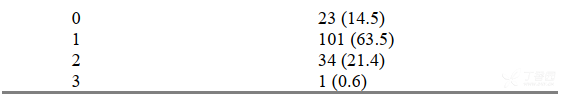
表12为疗效总结。在不同治疗亚组中客观缓解率显示有一定的差异性(根据入组时基线特征进行分组,受试者的客观缓解率情况见表13)。吉非替尼对这些亚组受试者的效果和预期的相一致。


遗传毒性:
吉非替尼体外试验(细菌、小鼠淋巴瘤和人淋巴细胞)和大鼠微核试验结果均为阴性。
生殖毒性:
在一项专门的大鼠生育力试验中,当吉非替尼的剂量≥120mg/2(按体表面积剂量计,与临床用药剂量相当)时,可见发情紊乱发生率升高、黄体数减少以及每窝活胎数和子宫着床数减少。
在大鼠妊娠及分娩期间经口给予吉非替尼20mg/kg/日(按体表面积剂量计,为临床用药剂量的0.7倍),幼鼠存活率降低。
在动物生殖毒性试验中,从器官形成期到离乳经口给予低于临床用药剂量的吉非替尼可引起胚胎毒性和新生动物死亡。
妊娠大鼠从器官形成期到离乳给予吉非替尼5mg/kg,存活新生鼠减少;剂量为20mg/kg(按体表面积剂量计,与临床剂量大致相当)时毒性更为严重,幼仔出生后死亡的比例增加。兔给予吉非替尼20mg/kg/日(240mg/2,按体表面积剂量计,约为临床用药剂量的2倍)胎仔体重降低。
大鼠单次经口给予吉非替尼5mg/kg(按体表面积剂量计,约为临床用药剂量的0.2倍),吉非替尼可透过胎盘;吉非替尼及某些代谢产物广泛分泌入乳汁。
致癌性:
在两年的小鼠致癌性试验中,当吉非替尼的剂量为270mg/2/日(自第22周开始由375mg/v/日减低至此剂量,按体表面积剂量计,约为临床用药剂量的2倍)时,雌性动物可见肝细胞腺瘤。
在两年的大鼠致癌性试验中,当吉非替尼的剂量为60mg/2/日(按体表面积剂量计,约为临床用药剂量的0.4倍)时,雌性动物可见肝细胞腺瘤和肠系膜淋巴结血管瘤/血管肉瘤。尚不了解这些发现的临床相关性。
化学名称:N-(3-氯-4-氟苯基)-7-甲氧基-6-(3-吗啉丙氧基)喹唑啉-4-胺
化学结构式:
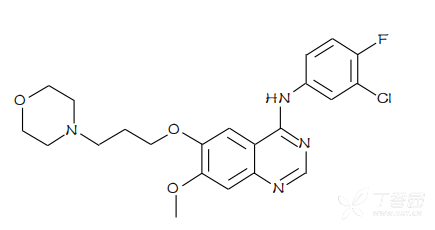
分子式:C22H24ClFN4O3
分子量:446.90



 购物车
购物车